 Les cardiopathies congénitales sont les anomalies cardiaques survenant au cours de la formation du cœur pendant la vie intra-utérine, c’est-à-dire les malformations du cœur du bébé qui se font avant la naissance. Elles sont très diverses, allant de la simple anomalie bénigne permettant la croissance de l’enfant sans aucun problème, jusqu’à la malformation grave, mettant en danger la survie du nouveau-né. Elles sont aussi très nombreuses et parfois très compliquées, ce qui nécessite obligatoirement de consulter des spécialistes. Jusqu’à quatorze fœtus sur mille sont porteurs d’une malformation cardiaque, faisant de ces dernières les malformations les plus fréquentes de l’enfant.
Les cardiopathies congénitales sont les anomalies cardiaques survenant au cours de la formation du cœur pendant la vie intra-utérine, c’est-à-dire les malformations du cœur du bébé qui se font avant la naissance. Elles sont très diverses, allant de la simple anomalie bénigne permettant la croissance de l’enfant sans aucun problème, jusqu’à la malformation grave, mettant en danger la survie du nouveau-né. Elles sont aussi très nombreuses et parfois très compliquées, ce qui nécessite obligatoirement de consulter des spécialistes. Jusqu’à quatorze fœtus sur mille sont porteurs d’une malformation cardiaque, faisant de ces dernières les malformations les plus fréquentes de l’enfant.
Le cœur normal
La formation du cœur commence 18 jours après la fécondation et se termine vers le 42ème jour après la fécondation.
Le risque
Les malformations cardiaques congénitales sont de 5.000 cas environ en France, soit 7 à 8 pour 1.000 naissances. Si une mère a déjà accouché d’un enfant porteur d’une malformation cardiaque, le risque passe de 1% à 5% environ et il passe à 10% si la mère a déjà accouché de deux enfants porteurs d’une malformation cardiaque. Une mère ayant elle-même une malformation cardiaque peut parfois avoir des enfants, mais elle a un risque augmenté de mettre au monde un enfant porteur aussi d’une malformation cardiaque.
 Un nouveau-né chez qui on trouve un souffle ou une anomalie à l’auscultation cardiaque ou encore une cyanose, est suspect de maladie cardiaque congénitale. Les cardiopathies cyanogènes sont des malformations cardiaques où il y a un mélange de sang non oxygéné (bleu) avec du sang oxygéné (rouge). Une coloration bleue-violacée du visage, des lèvres et des ongles apparaît, surtout lors de pleurs. Elle est due à une oxygénation insuffisante du sang d’où le nom de “maladie bleue” ou d’“enfant bleu”.
Un nouveau-né chez qui on trouve un souffle ou une anomalie à l’auscultation cardiaque ou encore une cyanose, est suspect de maladie cardiaque congénitale. Les cardiopathies cyanogènes sont des malformations cardiaques où il y a un mélange de sang non oxygéné (bleu) avec du sang oxygéné (rouge). Une coloration bleue-violacée du visage, des lèvres et des ongles apparaît, surtout lors de pleurs. Elle est due à une oxygénation insuffisante du sang d’où le nom de “maladie bleue” ou d’“enfant bleu”.
On fera d’autres examens comme la radiographie du cœur et des poumons qui peuvent montrer une augmentation du volume du cœur ou des anomalies des vaisseaux pulmonaires. On complètera par un électrocardiogramme, mais l’examen principal est l’échocardiographie qui a transformé le diagnostic des cardiopathies congénitales.
Une cause n’est retrouvée que dans 15% des cas environ, c’est-à-dire que dans la grande majorité des cas, on n’en trouve pas, d’où l’intérêt de faire un dépistage chez tout le monde.
Les causes génétiques représentent 8 à 10% des cas, avec en tête la trisomie 21 ou mongolisme. Il faut savoir que 40% des enfants trisomiques ont une anomalie cardiaque à la naissance. Les autres sont la trisomie 18 et le syndrome de Turner.
Certaines maladies virales maternelles comme la rubéole s’accompagnent dans 35% des cas de cardiopathies. C’est pourquoi, la vaccination des petites filles est nécessaire pour leur éviter d’attraper la rubéole lorsqu’elles grandissent et ont une grossesse. Pour celles qui n’ont pas été vaccinées, une sérologie en début de grossesse est systématique. Si elle est négative, une surveillance mensuelle sera faite pour vérifier l’absence de contamination. Si une femme a un diabète ou une maladie comme le lupus, le risque d’avoir un bébé avec une cardiopathie congénitale est plus élevé que la normale. De même, une consommation d’alcool pendant la grossesse augmente ce risque qui peut aller jusqu’à 25% dans les cas extrêmes.
Certains médicaments sont interdits chez la femme enceinte, car ils risquent de provoquer des anomalies chez le bébé. Il faut aussi se méfier des radiations ionisantes comme par exemple l’irradiation de Tchernobyl. Les radiographies et scanners sont interdits pendant la grossesse sauf cas très particuliers.
La communication inter ventriculaire appelée C.I.V. constitue 25% des maladies cardiaques congénitales. C’est un trou entre le cœur gauche et le cœur droit par lequel le sang va passer et se mélanger. Elle a pour conséquence un enrichissement en oxygène du sang des cavités droites et une augmentation du débit dans les artères pulmonaires, ce qui entraîne des difficultés à respirer et des surinfections. Elle peut aussi retarder la croissance avec un retard en poids et en taille par rapport aux valeurs normales pour l’âge. Une déformation du thorax avec un bombement vers l’avant peut parfois se produire.
 En fait, la C.I.V se présente sous plusieurs formes. La forme la plus bénigne est le type I. L’enfant ne se plaint de rien, mais on constate un souffle à l’examen. Dans le type II, le nourrisson ne grandit pas bien, il a du mal à boire ses biberons, car il est essoufflé et fait souvent des infections respiratoires. L’échographie montre la C.I.V. ou trou entre les deux ventricules, ainsi que son retentissement sur le reste du cœur et l’artère pulmonaire.
En fait, la C.I.V se présente sous plusieurs formes. La forme la plus bénigne est le type I. L’enfant ne se plaint de rien, mais on constate un souffle à l’examen. Dans le type II, le nourrisson ne grandit pas bien, il a du mal à boire ses biberons, car il est essoufflé et fait souvent des infections respiratoires. L’échographie montre la C.I.V. ou trou entre les deux ventricules, ainsi que son retentissement sur le reste du cœur et l’artère pulmonaire.
Le cathétérisme est souvent indispensable avant de décider d’une éventuelle intervention.
Le type II est une forme évoluée, souvent négligée et le type IV est appelé à “poumons protégés” car il s’accompagne d’un rétrécissement de l’artère pulmonaire.
Les points importants à prendre en compte sont la taille de la C.I.V. et de l’existence ou non d’une sténose pulmonaire.
En général, le type I ne pose pas de problèmes. Le trou peut soit se fermer spontanément ou rester petit sans conséquences.
Le type II peut aussi se fermer spontanément ou rester stable ou évoluer vers une forme plus grave. C’est celui qu’il faut surveiller de près pour décider du bon moment pour une éventuelle chirurgie ou fermeture percutanée par une ombrelle. La chirurgie se fait à cœur ouvert en mettant un matériel chirurgical appelé patch pour recouvrir le trou. L’ombrelle est une sorte de petit parapluie de métal qu’on met en cours de cathétérisme, sans ouvrir le thorax et donc sans chirurgie. Le choix entre les deux techniques dépend de plusieurs facteurs mesurés lors de l’échographie et du cathétérisme cardiaque. On réalise ces interventions avant que les résistances pulmonaires ne soient élevées de façon irréversible, c’est-à-dire en général avant l’âge de 2 ans.
Sous l’acronyme C.A.V., il est plus graveque les deux autres. Il associe en fait C.I.A. et C.I.V. large. Il s’observe fréquemment dans la trisomie 21, il représente la moitié des cardiopathies des enfants trisomiques. Son traitement est chirurgicale et il s’agit d’une chirurgie difficile.
 Elle représente 8% des malformations cardiaques congénitales et c’est la plus fréquente des “maladies bleues”. La coloration bleue des lèvres et des ongles apparaît plus ou moins tôt souvent vers 3-6 mois, mais parfois plus tardivement. Les complications les plus spectaculaires sont les crises d’anoxie: accentuation brutale de la cyanose, avec malaise, baisse de tonus et perte de connaissance. C’est l’échographie cardiaque qui affirme le diagnostic. Le traitement ne peut être que chirurgical, idéalement ce qu’on appelle une cure complète qui répare toutes les anomalies. Mais cela n’est pas toujours possible et il faudra recourir à plusieurs opérations avant d’y arriver.
Elle représente 8% des malformations cardiaques congénitales et c’est la plus fréquente des “maladies bleues”. La coloration bleue des lèvres et des ongles apparaît plus ou moins tôt souvent vers 3-6 mois, mais parfois plus tardivement. Les complications les plus spectaculaires sont les crises d’anoxie: accentuation brutale de la cyanose, avec malaise, baisse de tonus et perte de connaissance. C’est l’échographie cardiaque qui affirme le diagnostic. Le traitement ne peut être que chirurgical, idéalement ce qu’on appelle une cure complète qui répare toutes les anomalies. Mais cela n’est pas toujours possible et il faudra recourir à plusieurs opérations avant d’y arriver.
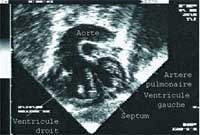 C’est la cardiopathie congénitale cyanogène la plus fréquente en période néonatale. C’est l’autre forme la plus connue de ce qu’on appelait “enfant bleu”. Dans cette malformation cardiaque, les deux gros vaisseaux qui sortent du cœur et qui sont l’aorte et l’artère pulmonaire, sont inversés. Elle est définie par la naissance de l’aorte à partir du ventricule droit et de l’artère pulmonaire à partir du ventricule gauche, ce qui est le contraire de la normale.
C’est la cardiopathie congénitale cyanogène la plus fréquente en période néonatale. C’est l’autre forme la plus connue de ce qu’on appelait “enfant bleu”. Dans cette malformation cardiaque, les deux gros vaisseaux qui sortent du cœur et qui sont l’aorte et l’artère pulmonaire, sont inversés. Elle est définie par la naissance de l’aorte à partir du ventricule droit et de l’artère pulmonaire à partir du ventricule gauche, ce qui est le contraire de la normale.
Elle se révèle dès les tout premiers jours de la vie par une cyanose intense qui ne se corrige pas sous oxygène. L’échographie cardiaque montre un croisement des deux vaisseaux qui se chevauchent. Très souvent, en raison de la cyanose importante et de l’apparition d’une acidose, elle nécessite en urgence ou en semi urgence un geste de cathétérisme interventionnel appelé manœuvre de Rahskind qui permet de tenir quelques jours pour mieux préparer la chirurgie.
 C’est la sténose ou rétrécissement anormal de la partie initiale de l’aorte descendante, peu après sa sortie du cœur. Elle peut apparaître tôt chez le nourrisson, mais le plus souvent un peu plus tard chez le jeune enfant. Chez le nourrisson, c’est la forme la plus aiguë qui se révèle le plus souvent dans les premières semaines de la vie par une insuffisance cardiaque. Cette anomalie empêche le sang de bien arriver au reste du corps et on ne trouve pas chez le bébé de pulsatilité des pouls fémoraux, ce qui évoque très fortement le diagnostic. Chez le jeune enfant, on trouvera un souffle systolique, un pouls fémoral très faible ou inexistant et une hypertension aux deux bras.
C’est la sténose ou rétrécissement anormal de la partie initiale de l’aorte descendante, peu après sa sortie du cœur. Elle peut apparaître tôt chez le nourrisson, mais le plus souvent un peu plus tard chez le jeune enfant. Chez le nourrisson, c’est la forme la plus aiguë qui se révèle le plus souvent dans les premières semaines de la vie par une insuffisance cardiaque. Cette anomalie empêche le sang de bien arriver au reste du corps et on ne trouve pas chez le bébé de pulsatilité des pouls fémoraux, ce qui évoque très fortement le diagnostic. Chez le jeune enfant, on trouvera un souffle systolique, un pouls fémoral très faible ou inexistant et une hypertension aux deux bras.
L’échocardiographie montrera la coarctation dans la plupart des cas. Si on ne fait rien, le risque est l’évolution vers une hypertension artérielle et ses complications, notamment les accidents vasculaires cérébraux. Le traitement classique est l’intervention chirurgicale qui consiste le plus souvent à couper la zone malade et re suturer les deux morceaux sains bout à bout dans la région de l’isthme aortique.
 Les cardiopathies congénitales sont des affections relativement fréquentes: 8 pour 1.000 naissances. Cette fréquence diminue ensuite avec l’âge car un certain nombre d’enfants, de moins en moins nombreux de nos jours, porteurs de cardiopathies inopérables, vont mourir, généralement jeunes. Par contre, certaines anomalies mineures présentes à la naissance vont rapidement disparaître spontanément sans nécessiter d’opération.
Les cardiopathies congénitales sont des affections relativement fréquentes: 8 pour 1.000 naissances. Cette fréquence diminue ensuite avec l’âge car un certain nombre d’enfants, de moins en moins nombreux de nos jours, porteurs de cardiopathies inopérables, vont mourir, généralement jeunes. Par contre, certaines anomalies mineures présentes à la naissance vont rapidement disparaître spontanément sans nécessiter d’opération.
Le dépistage des maladies cardiaques congénitales est un objectif principal de l’échographie morphologique du fœtus.
Le diagnostic permettra de faire des examens complémentaires, comme une amniocentèse pour la recherche d’une trisomie 21.
La présence de certaines cardiopathies nécessite l’accouchement de la mère dans une maternité spécialisée pour une prise en charge du nouveau-né.